Use-case: Exploring perturbation effects#
This tutorial shows how to perform the analysis we present in Figure 4 of the pertpy preprint. We will use the data from Zhang et al. 2021 to explore the perturbation effects of different treatments on triple-negative breast cancer. We will use the mean squared error (MSE) distance to compare the expression profiles of cells between the different treatment-response groups. Additionally, we will use scCODA to identify changes in cell type composition between the different treatment groups.
import warnings
warnings.filterwarnings("ignore")
import matplotlib.pyplot as plt
import pandas as pd
import pertpy as pt
import scanpy as sc
import seaborn as sns
Load and preprocess the data#
adata = pt.dt.zhang_2021()
adata
AnnData object with n_obs × n_vars = 489490 × 27085
obs: 'Sample', 'Patient', 'Origin', 'Tissue', 'Efficacy', 'Group', 'Treatment', 'Number of counts', 'Number of genes', 'Major celltype', 'Cluster'
First, we will filter the data to only include tumor samples, no progression samples, and only samples with a partial response or stable disease. We will also filter out the samples with a mix of cell types, following the pre-processing steps in the original publication.
# Filter to tumor samples only
adata = adata[adata.obs["Origin"] == "t", :].copy()
# Filter out progression samples
adata = adata[adata.obs["Group"] != "Progression", :].copy()
# Subset to partial response and stable disease and rename PR and SD
adata = adata[adata.obs["Efficacy"].isin(["PR", "SD"]), :].copy()
adata.obs["Efficacy"] = adata.obs["Efficacy"].replace({"PR": "Partial response", "SD": "Stable disease"})
# Filter out Mix samples
adata = adata[adata.obs["Cluster"] != "Mix", :].copy()
adata
AnnData object with n_obs × n_vars = 146358 × 27085
obs: 'Sample', 'Patient', 'Origin', 'Tissue', 'Efficacy', 'Group', 'Treatment', 'Number of counts', 'Number of genes', 'Major celltype', 'Cluster'
Now, we will refine the grouping to include both the timepoint and the treatment response.
adata.obs["Timepoint"] = adata.obs["Group"].copy()
adata.obs["Group"] = [
f"{timepoint.split('-')[0]}-treat., {response}"
for timepoint, response in zip(adata.obs["Timepoint"], adata.obs["Efficacy"])
]
adata.obs["Group"].value_counts()
Group
Pre-treat., Partial response 57295
Post-treat., Stable disease 31626
Post-treat., Partial response 29659
Pre-treat., Stable disease 27778
Name: count, dtype: int64
After filtering, we do some standard pre-processing steps:
sc.pp.filter_genes(adata, min_cells=10)
sc.pp.normalize_total(adata, target_sum=1e4)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata, n_top_genes=4000, flavor="seurat_v3")
adata.raw = adata
adata = adata[:, adata.var.highly_variable]
adata
View of AnnData object with n_obs × n_vars = 146358 × 4000
obs: 'Sample', 'Patient', 'Origin', 'Tissue', 'Efficacy', 'Group', 'Treatment', 'Number of counts', 'Number of genes', 'Major celltype', 'Cluster', 'Timepoint'
var: 'n_cells', 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm'
uns: 'log1p', 'hvg'
sc.pp.scale(adata, max_value=10)
sc.tl.pca(adata, svd_solver="arpack")
sc.pp.neighbors(adata)
sc.tl.umap(adata)
sc.pl.umap(adata, color="Treatment")
sc.pl.umap(adata, color="Group")
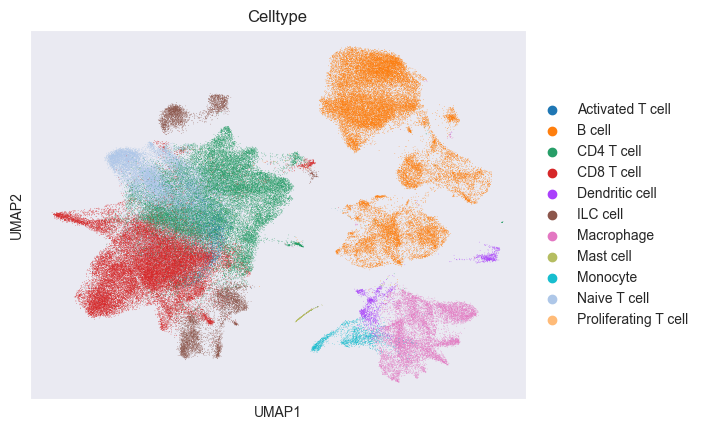
The dataset comes with a very detailed cell type annotation. We will use this annotation to define a coarser cell type annotation:
cell_type_converter = {
"t_Bfoc": "B cell",
"t_Bmem": "B cell",
"t_Bn": "B cell",
"t_CD4": "CD4 T cell",
"t_CD8": "CD8 T cell",
"t_ILC": "ILC cell",
"t_Tact": "Activated T cell",
"t_Tn": "Naive T cell",
"t_Tprf": "Proliferating T cell",
"t_cDC": "Dendritic cell",
"t_mDC": "Dendritic cell",
"t_macro": "Macrophage",
"t_mast": "Mast cell",
"t_mono": "Monocyte",
"t_pB": "B cell",
"t_pDC": "Dendritic cell",
}
cell_types = []
for ct in adata.obs["Cluster"]:
for key, value in cell_type_converter.items():
if ct.startswith(key):
cell_types.append(value)
break
else:
cell_types.append("n.a.")
adata.obs["Celltype"] = cell_types
sc.pl.umap(adata, color="Celltype")
We will further define a function to filter the data to only include cell types that are present in all groups. This function is used to create adatas for the different treatment groups. We will use those for the distance metric analysis, as well as for the compositional analysis.
def subset_to_common_cell_types(adata_temp):
isecs = pd.crosstab(adata_temp.obs["Cluster"], adata_temp.obs["Group"])
celltypes = isecs[(isecs > 0).all(axis=1)].index.values.tolist()
adata_temp = adata_temp[adata_temp.obs["Cluster"].isin(celltypes)]
return adata_temp
adata_chemo = adata[adata.obs["Treatment"] == "Chemo"]
adata_chemo = subset_to_common_cell_types(adata_chemo)
adata_chemo.obs["Group"].value_counts()
Group
Post-treat., Stable disease 20659
Pre-treat., Partial response 16827
Post-treat., Partial response 12137
Pre-treat., Stable disease 11807
Name: count, dtype: int64
adata_chemo_pdl1 = adata[adata.obs["Treatment"] == "Anti-PD-L1+Chemo"]
adata_chemo_pdl1 = subset_to_common_cell_types(adata_chemo_pdl1)
adata_chemo_pdl1.obs["Group"].value_counts()
Group
Pre-treat., Partial response 40251
Post-treat., Partial response 16379
Pre-treat., Stable disease 13435
Post-treat., Stable disease 10717
Name: count, dtype: int64
Using a distance metric to rank perturbation effects#
Pertpy offers various distance metrics to compare the expression profile of cells between different groups, usually different perturbations. Here, we will use the mean squared error (MSE) distance to compare the expression profiles of cells between the different treatment-response groups. Importantly, we will do this for the full dataset, as well as for the chemo and chemo + anti-PD-L1 groups separately. This allows us to identify potential differences in the perturbation effects between the two treatment groups.
distance = pt.tl.Distance("mse", obsm_key="X_pca")
df_all = distance.pairwise(adata, groupby="Group", show_progressbar=False)
df_chemo = distance.pairwise(adata_chemo, groupby="Group", show_progressbar=False)
df_chemo_pdl1 = distance.pairwise(adata_chemo_pdl1, groupby="Group", show_progressbar=False)
# We need the global max and min to ensure that the color scale is the same for all heatmaps
global_max = max(df_all.max(axis=None), df_chemo.max(axis=None), df_chemo_pdl1.max(axis=None))
global_min = min(df_all.min(axis=None), df_chemo.min(axis=None), df_chemo_pdl1.min(axis=None))
order = df_all.index.values
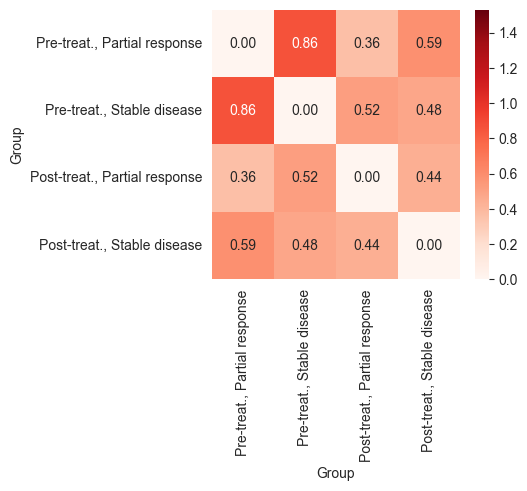
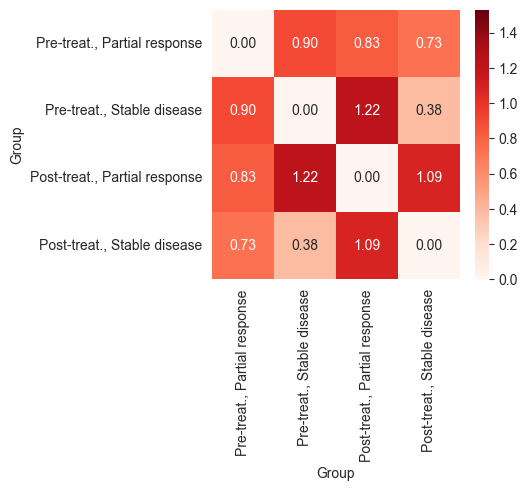
_, ax = plt.subplots(figsize=(4, 3.5))
sns.heatmap(df_all, annot=True, fmt=".2f", vmin=global_min, vmax=global_max, cmap="Reds", ax=ax)
plt.show()
df_chemo = df_chemo.loc[order, order]
_, ax = plt.subplots(figsize=(4, 3.5))
sns.heatmap(df_chemo, annot=True, fmt=".2f", vmin=global_min, vmax=global_max, cmap="Reds", ax=ax)
plt.show()
df_chemo_pdl1 = df_chemo_pdl1.loc[order, order]
_, ax = plt.subplots(figsize=(4, 3.5))
sns.heatmap(df_chemo_pdl1, annot=True, fmt=".2f", vmin=global_min, vmax=global_max, cmap="Reds", ax=ax)
plt.show()
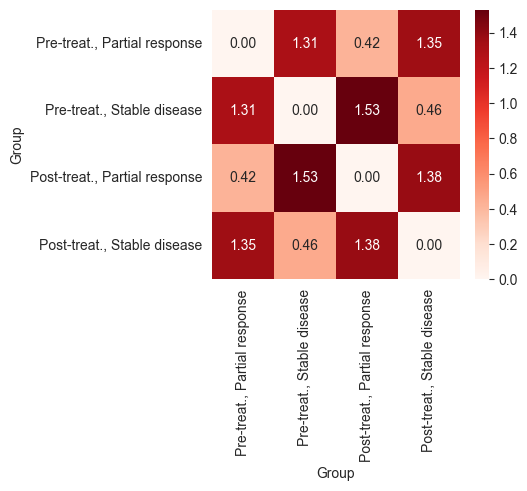
The heatmaps above show that patients who responded to chemotherapy showed a larger difference between their pre- and post-treatment expression profiles compared to those who responded to the combination of anti-PDL-1 and chemotherapy. This indicates that the combination therapy may have led to a less intense response or was applied in cases with poorer prognoses.
Identifying changes in cell type composition using scCODA#
scCODA is a Bayesian model that allows to identify changes in cell type composition. Here, we will use scCODA to identify changes in cell type composition between the different treatment groups. We will run, analogous to the analysis set-up for the distance metric calculation, scCODA for each treatment (Chemo and Anti-PD-L1+Chemo) separately. However, we want to use the same reference cell type in order to be able to compare the results. Hence, we will first prepare the scCODA model on all data to identify a reference cell type:
# Get reference cell type
sccoda_model = pt.tl.Sccoda()
sccoda_data = sccoda_model.load(
adata,
type="cell_level",
generate_sample_level=True,
cell_type_identifier="Cluster",
sample_identifier="Sample",
covariate_obs=["Group"],
)
sccoda_data = sccoda_model.prepare(
sccoda_data,
modality_key="coda",
formula="Group",
reference_cell_type="automatic",
automatic_reference_absence_threshold=0.1,
)
💡 Automatic reference selection! Reference cell type set to t_mono-FCN1
💡 Zero counts encountered in data! Added a pseudocount of 0.5.
fig = sccoda_model.plot_boxplots(
sccoda_data,
modality_key="coda",
feature_name="Group",
return_fig=True,
show=False,
)
fig.set_size_inches(25, 5)
fig.set_dpi(200)
fig.show()
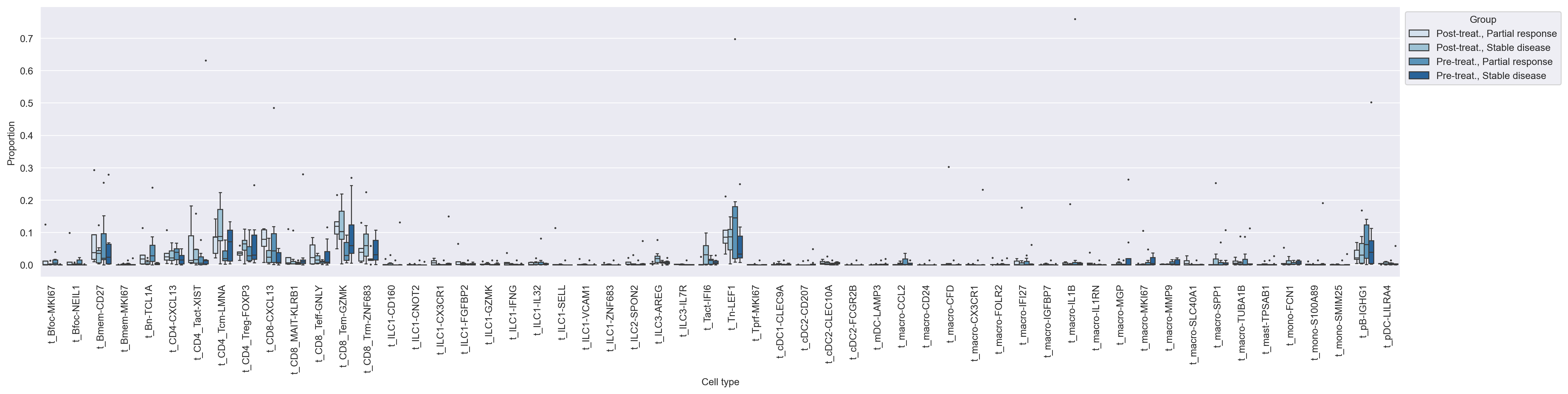
The monocyte cell type is identified as the reference cell type. We will now run scCODA for the chemo and chemo + anti-PD-L1 groups separately, using the monocyte cell type as the reference cell type.
sccoda_model = pt.tl.Sccoda()
def run_sccoda(subset, reference):
sccoda_data = sccoda_model.load(
subset,
type="cell_level",
generate_sample_level=True,
cell_type_identifier="Cluster",
sample_identifier="Sample",
covariate_obs=["Group"],
)
sccoda_data = sccoda_model.prepare(
sccoda_data,
modality_key="coda",
formula=f"C(Group, Treatment('{reference}'))",
reference_cell_type="t_mono-FCN1", # "automatic",
automatic_reference_absence_threshold=0.1,
)
sccoda_model.run_nuts(sccoda_data, modality_key="coda")
sccoda_model.set_fdr(sccoda_data, modality_key="coda", est_fdr=0.1)
comparison_groups = [g for g in subset.obs["Group"].unique() if g != reference]
effect_df = pd.DataFrame(
{"log2-fold change": [], "Cell Type": [], "Reference": [], "Comp. Group": [], "Final Parameter": []}
)
for comp_group in comparison_groups:
group_effects = sccoda_data["coda"].varm[f"effect_df_C(Group, Treatment('{reference}'))[T.{comp_group}]"][
["log2-fold change", "Final Parameter"]
]
group_effects = group_effects[group_effects["Final Parameter"] != 0]
group_effects["Cell Type"] = group_effects.index
group_effects["Reference"] = reference
group_effects["Comp. Group"] = comp_group
effect_df = pd.concat([effect_df, group_effects])
if not effect_df.empty:
fig = sccoda_model.plot_effects_barplot(sccoda_data, return_fig=True, show=False)
fig.set_size_inches(12, 4)
fig.show()
else:
print(f"No significant effects for reference {reference}")
return effect_df
credible_effects_chemo = pd.DataFrame(
{"log2-fold change": [], "Cell Type": [], "Reference": [], "Comp. Group": [], "Final Parameter": []}
)
for reference in adata_chemo.obs["Group"].unique():
print(reference)
effect_df_chemo = run_sccoda(adata_chemo, reference=reference)
credible_effects_chemo = pd.concat([credible_effects_chemo, effect_df_chemo])
Pre-treat., Stable disease
💡 Zero counts encountered in data! Added a pseudocount of 0.5.
Pre-treat., Partial response
💡 Zero counts encountered in data! Added a pseudocount of 0.5.
No significant effects for reference Pre-treat., Partial response
Post-treat., Stable disease
💡 Zero counts encountered in data! Added a pseudocount of 0.5.
Post-treat., Partial response
💡 Zero counts encountered in data! Added a pseudocount of 0.5.
sample: 100%|██████████| 11000/11000 [02:14<00:00, 81.75it/s, 127 steps of size 2.63e-02. acc. prob=0.89]
sample: 100%|██████████| 11000/11000 [02:17<00:00, 80.06it/s, 127 steps of size 3.40e-02. acc. prob=0.85]
sample: 100%|██████████| 11000/11000 [02:19<00:00, 79.04it/s, 127 steps of size 2.78e-02. acc. prob=0.87]
sample: 100%|██████████| 11000/11000 [02:09<00:00, 84.70it/s, 127 steps of size 3.30e-02. acc. prob=0.79]
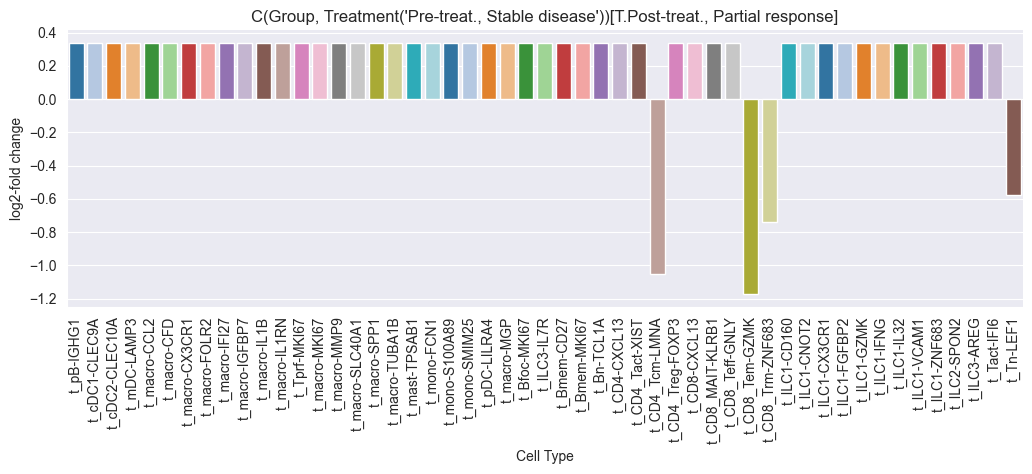
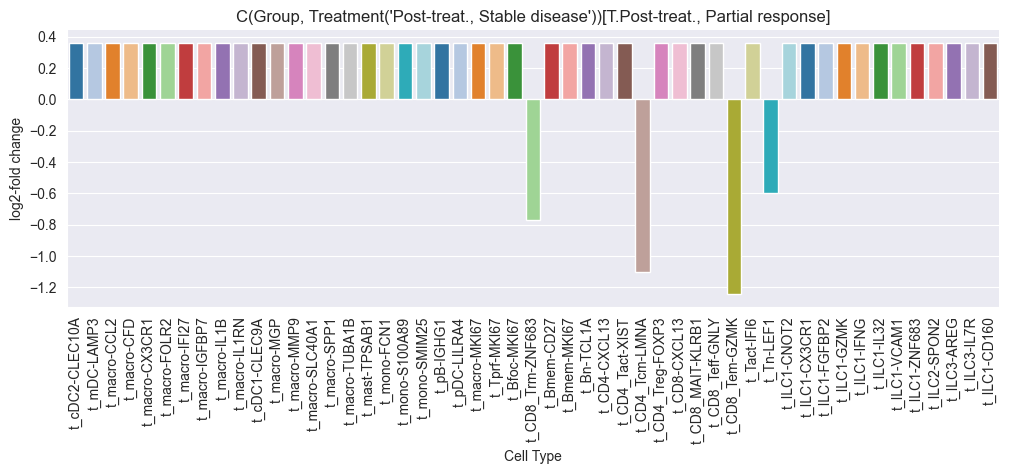
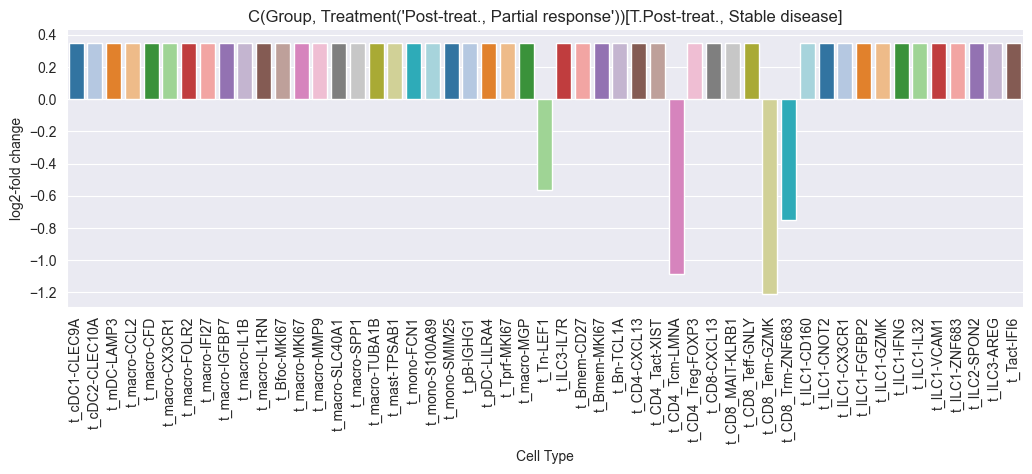
credible_effects_chemo
| log2-fold change | Cell Type | Reference | Comp. Group | Final Parameter | |
|---|---|---|---|---|---|
| t_CD4_Tcm-LMNA | -1.051869 | t_CD4_Tcm-LMNA | Pre-treat., Stable disease | Pre-treat., Partial response | -0.963443 |
| t_CD8_Tem-GZMK | -1.173683 | t_CD8_Tem-GZMK | Pre-treat., Stable disease | Pre-treat., Partial response | -1.047878 |
| t_CD8_Trm-ZNF683 | -0.737895 | t_CD8_Trm-ZNF683 | Pre-treat., Stable disease | Pre-treat., Partial response | -0.745813 |
| t_Tn-LEF1 | -0.577472 | t_Tn-LEF1 | Pre-treat., Stable disease | Pre-treat., Partial response | -0.634616 |
| t_CD4_Tcm-LMNA | -1.099619 | t_CD4_Tcm-LMNA | Post-treat., Stable disease | Pre-treat., Partial response | -1.012419 |
| t_CD8_Tem-GZMK | -1.243750 | t_CD8_Tem-GZMK | Post-treat., Stable disease | Pre-treat., Partial response | -1.112323 |
| t_CD8_Trm-ZNF683 | -0.768398 | t_CD8_Trm-ZNF683 | Post-treat., Stable disease | Pre-treat., Partial response | -0.782834 |
| t_Tn-LEF1 | -0.599843 | t_Tn-LEF1 | Post-treat., Stable disease | Pre-treat., Partial response | -0.666001 |
| t_CD4_Tcm-LMNA | -1.084423 | t_CD4_Tcm-LMNA | Post-treat., Partial response | Pre-treat., Partial response | -0.994157 |
| t_CD8_Tem-GZMK | -1.210772 | t_CD8_Tem-GZMK | Post-treat., Partial response | Pre-treat., Partial response | -1.081735 |
| t_CD8_Trm-ZNF683 | -0.748751 | t_CD8_Trm-ZNF683 | Post-treat., Partial response | Pre-treat., Partial response | -0.761486 |
| t_Tn-LEF1 | -0.566308 | t_Tn-LEF1 | Post-treat., Partial response | Pre-treat., Partial response | -0.635026 |
Multiple credible effects were found for the chemo treatment group. We will now run scCODA for the Anti-PD-L1 + Chemo group.
credible_effects_chemo_pdl1 = pd.DataFrame(
{"log2-fold change": [], "Cell Type": [], "Reference": [], "Comp. Group": [], "Final Parameter": []}
)
for reference in adata_chemo_pdl1.obs["Group"].unique():
print(reference)
effect_df_chemo_pdl1 = run_sccoda(adata_chemo_pdl1, reference=reference)
credible_effects_chemo_pdl1 = pd.concat([credible_effects_chemo_pdl1, effect_df_chemo_pdl1])
Pre-treat., Partial response
💡 Zero counts encountered in data! Added a pseudocount of 0.5.
No significant effects for reference Pre-treat., Partial response
Pre-treat., Stable disease
💡 Zero counts encountered in data! Added a pseudocount of 0.5.
No significant effects for reference Pre-treat., Stable disease
Post-treat., Partial response
💡 Zero counts encountered in data! Added a pseudocount of 0.5.
No significant effects for reference Post-treat., Partial response
Post-treat., Stable disease
💡 Zero counts encountered in data! Added a pseudocount of 0.5.
No significant effects for reference Post-treat., Stable disease
sample: 100%|██████████| 11000/11000 [02:08<00:00, 85.74it/s, 127 steps of size 4.46e-02. acc. prob=0.71]
sample: 100%|██████████| 11000/11000 [02:12<00:00, 82.78it/s, 127 steps of size 3.73e-02. acc. prob=0.77]
sample: 100%|██████████| 11000/11000 [02:09<00:00, 84.65it/s, 127 steps of size 3.12e-02. acc. prob=0.92]
sample: 100%|██████████| 11000/11000 [02:07<00:00, 86.24it/s, 127 steps of size 3.74e-02. acc. prob=0.75]
credible_effects_chemo_pdl1
| log2-fold change | Cell Type | Reference | Comp. Group | Final Parameter |
|---|
For the Anti-PD-L1 + Chemo treatment group, no credible effects, i.e. changes in cell type composition, were found.
This fits to our earlier findings from the distance metric analysis, where we observed that the Anti-PD-L1 + Chemo group showed a smaller difference between their pre- and post-treatment expression profiles compared to the Chemo group. Overall, these findings indicate that the Anti-PD-L1 + Chemo combination therapy may have led to a less intense response or was applied in cases with poorer prognoses.