pertpy.tools.PermutationTest#
- class PermutationTest(adata, *, mask=None, layer=None, **kwargs)[source]#
Perform a permutation test.
The permutation test relies on another test statistic (e.g. t-statistic or your own) to obtain a p-value through random permutations of the data and repeated generation of the test statistic.
For paired tests, each paired observation is permuted together and distributed randomly between the two groups. For unpaired tests, all observations are permuted independently.
The null hypothesis for the unpaired test is that all observations come from the same underlying distribution and have been randomly assigned to one of the samples.
The null hypothesis for the paired permutation test is that the observations within each pair are drawn from the same underlying distribution and that their assignment to a sample is random.
Attributes table#
Get the data matrix from anndata this object was initalized with (X or layer). |
Methods table#
|
Perform a permutation test comparison between groups. |
|
Plot a metric from the results as a bar chart, optionally with additional information about paired samples in a scatter plot. |
|
Plot a matrix of log2 fold changes from the results. |
|
Creates a pairwise expression plot from a Pandas DataFrame or Anndata. |
|
Creates a volcano plot from a pandas DataFrame or Anndata. |
Attributes#
- PermutationTest.data#
Get the data matrix from anndata this object was initalized with (X or layer).
Methods#
- classmethod PermutationTest.compare_groups(adata, column, baseline, groups_to_compare, *, paired_by=None, mask=None, layer=None, n_permutations=1000, test_statistic=<function PermutationTest.<lambda>>, fit_kwargs=mappingproxy({}), test_kwargs=mappingproxy({}))[source]#
Perform a permutation test comparison between groups.
- Parameters:
adata (
AnnData) – Data with observations to compare.column (
str) – Column in adata.obs that contains the groups to compare.baseline (
str) – Reference group.groups_to_compare (
str|Sequence[str]) – Groups to compare against the baseline. If None, all other groups are compared.paired_by (
str|None, default:None) – Column in adata.obs to use for pairing. If None, an unpaired test is performed.mask (
str|None, default:None) – Mask to apply to the data.layer (
str|None, default:None) – Layer to use for the comparison.n_permutations (
int, default:1000) – Number of permutations to perform.test_statistic (
Callable[[ndarray,ndarray],float], default:<function PermutationTest.<lambda> at 0x7017f3419bc0>) – A callable that takes two arrays (x0, x1) and returns a float statistic. Defaults to log2 fold change with pseudocount: log2(mean(x1) + 1e-8) - log2(mean(x0) + 1e-8). The callable should have signature: test_statistic(x0, x1) -> float.fit_kwargs (
Mapping, default:mappingproxy({})) – Unused argument for compatibility with the MethodBase interface, do not specify.test_kwargs (
Mapping, default:mappingproxy({})) – Additional kwargs passed to the permutation test function (not the test statistic). The permutation test function is scipy.stats.permutation_test. We refer to its documentation for available options. Note that test_statistic and n_permutations are set by this function and should not be provided here.
- Return type:
Examples
>>> # Difference in means (log fold change) >>> PermutationTest.compare_groups( ... adata, ... column="condition", ... baseline="A", ... groups_to_compare="B", ... test_statistic=lambda x, y: np.log2(np.mean(y)) - np.log2(np.mean(x)), ... n_permutations=1000, ... test_kwargs={"rng": 0}, ... )
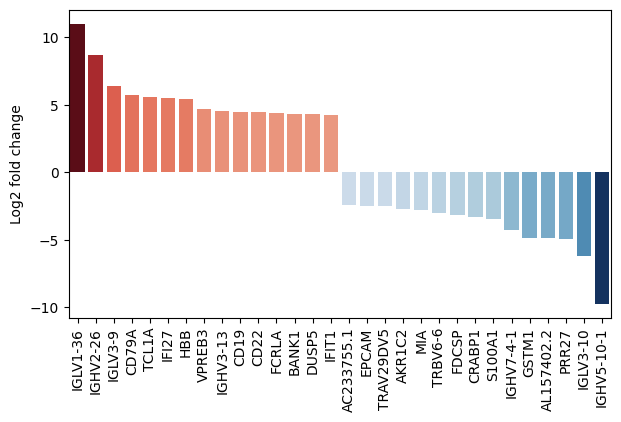
- PermutationTest.plot_fold_change(results_df, *, var_names=None, n_top_vars=15, log2fc_col='log_fc', symbol_col='variable', y_label='Log2 fold change', figsize=(10, 5), return_fig=False, **barplot_kwargs)#
Plot a metric from the results as a bar chart, optionally with additional information about paired samples in a scatter plot.
- Parameters:
results_df (
DataFrame) – DataFrame with results from DE analysis.var_names (
Sequence[str], default:None) – Variables to plot. If None, the top n_top_vars variables based on the log2 fold change are plotted.n_top_vars (
int, default:15) – Number of top variables to plot. The top and bottom n_top_vars variables are plotted, respectively.log2fc_col (
str, default:'log_fc') – Column name of log2 Fold-Change values.symbol_col (
str, default:'variable') – Column name of gene IDs.y_label (
str, default:'Log2 fold change') – Label for the y-axis.figsize (
tuple[int,int], default:(10, 5)) – Size of the figure.return_fig (
bool, default:False) – if True, returns figure of the plot, that can be used for saving.**barplot_kwargs – Additional arguments for seaborn.barplot.
- Return type:
- Returns:
If return_fig is True, returns the figure, otherwise None.
Examples
>>> # Example with EdgeR >>> import pertpy as pt >>> adata = pt.dt.zhang_2021() >>> adata.layers["counts"] = adata.X.copy() >>> ps = pt.tl.PseudobulkSpace() >>> pdata = ps.compute( ... adata, ... target_col="Patient", ... groups_col="Cluster", ... layer_key="counts", ... mode="sum", ... min_cells=10, ... min_counts=1000, ... ) >>> edgr = pt.tl.EdgeR(pdata, design="~Efficacy+Treatment") >>> edgr.fit() >>> res_df = edgr.test_contrasts( ... edgr.contrast(column="Treatment", baseline="Chemo", group_to_compare="Anti-PD-L1+Chemo") ... ) >>> edgr.plot_fold_change(res_df)
- Preview:
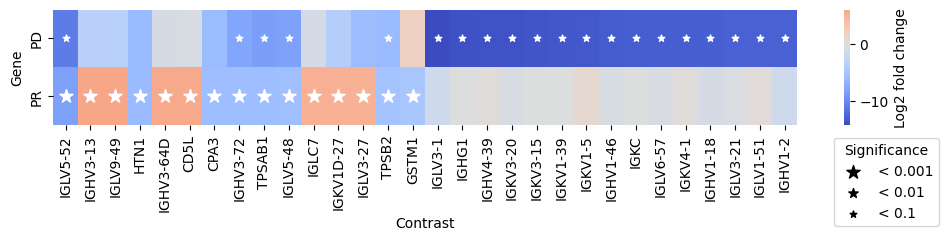
- PermutationTest.plot_multicomparison_fc(results_df, *, n_top_vars=15, contrast_col='contrast', log2fc_col='log_fc', pvalue_col='adj_p_value', symbol_col='variable', marker_size=100, figsize=(10, 2), x_label='Contrast', y_label='Gene', return_fig=False, **heatmap_kwargs)#
Plot a matrix of log2 fold changes from the results.
- Parameters:
results_df (
DataFrame) – DataFrame with results from DE analysis.n_top_vars (default:
15) – Number of top variables to plot per group.contrast_col (
str, default:'contrast') – Column in results_df containing information about the contrast.log2fc_col (
str, default:'log_fc') – Column in results_df containing the log2 fold change.pvalue_col (
str, default:'adj_p_value') – Column in results_df containing the p-value. Can be used to switch between adjusted and unadjusted p-values.symbol_col (
str, default:'variable') – Column in results_df containing the gene symbol.marker_size (
int, default:100) – Size of the biggest marker for significant variables.figsize (
tuple[int,int], default:(10, 2)) – Size of the figure.x_label (
str, default:'Contrast') – Label for the x-axis.y_label (
str, default:'Gene') – Label for the y-axis.return_fig (
bool, default:False) – if True, returns figure of the plot, that can be used for saving.**heatmap_kwargs – Additional arguments for seaborn.heatmap.
- Return type:
- Returns:
If return_fig is True, returns the figure, otherwise None.
Examples
>>> # Example with EdgeR >>> import pertpy as pt >>> adata = pt.dt.zhang_2021() >>> adata.layers["counts"] = adata.X.copy() >>> ps = pt.tl.PseudobulkSpace() >>> pdata = ps.compute( ... adata, ... target_col="Patient", ... groups_col="Cluster", ... layer_key="counts", ... mode="sum", ... min_cells=10, ... min_counts=1000, ... ) >>> edgr = pt.tl.EdgeR(pdata, design="~Efficacy+Treatment") >>> res_df = edgr.compare_groups(pdata, column="Efficacy", baseline="SD", groups_to_compare=["PR", "PD"]) >>> edgr.plot_multicomparison_fc(res_df)
- Preview:
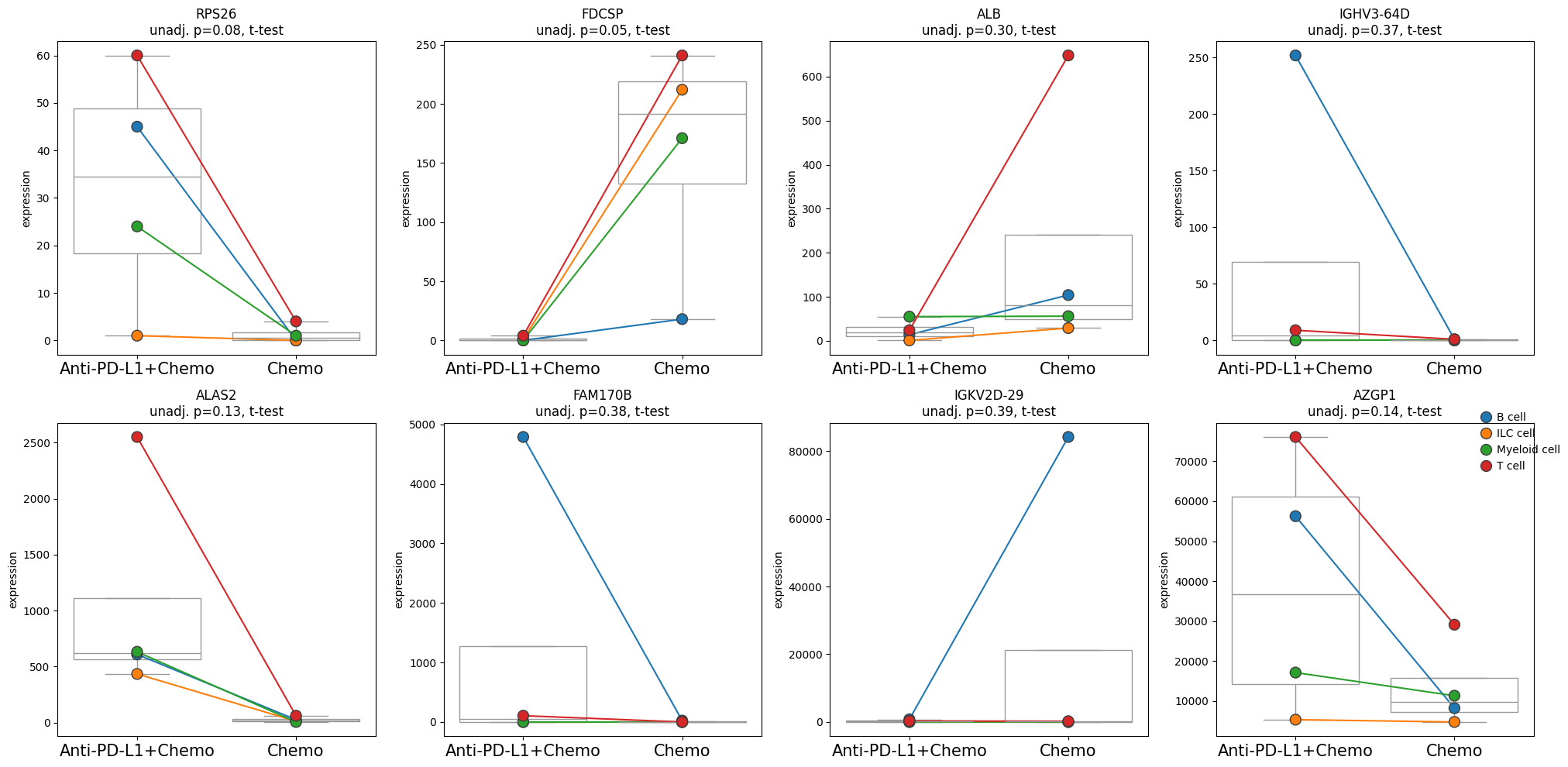
- PermutationTest.plot_paired(adata, results_df, groupby, pairedby, *, var_names=None, n_top_vars=15, layer=None, pvalue_col='adj_p_value', symbol_col='variable', n_cols=4, panel_size=(5, 5), show_legend=True, size=10, y_label='expression', pvalue_template=<function MethodBase.<lambda>>, boxplot_properties=None, palette=None, return_fig=False)#
Creates a pairwise expression plot from a Pandas DataFrame or Anndata.
Visualizes a panel of paired scatterplots per variable.
- Parameters:
adata (
AnnData) – AnnData object, can be pseudobulked.results_df (
DataFrame) – DataFrame with results from a differential expression test.groupby (
str) – .obs column containing the grouping. Must contain exactly two different values.pairedby (
str) – .obs column containing the pairing (e.g. “patient_id”). If None, an independent t-test is performed.var_names (
Sequence[str], default:None) – Variables to plot.n_top_vars (
int, default:15) – Number of top variables to plot.layer (
str, default:None) – Layer to use for plotting.pvalue_col (
str, default:'adj_p_value') – Column name of the p values.symbol_col (
str, default:'variable') – Column name of gene IDs.n_cols (
int, default:4) – Number of columns in the plot.panel_size (
tuple[int,int], default:(5, 5)) – Size of each panel.show_legend (
bool, default:True) – Whether to show the legend.size (
int, default:10) – Size of the points.y_label (
str, default:'expression') – Label for the y-axis.pvalue_template (default:
<function MethodBase.<lambda> at 0x7017f34189a0>) – Template for the p-value string displayed in the title of each panel.boxplot_properties (default:
None) – Additional properties for the boxplot, passed to seaborn.boxplot.palette (default:
None) – Color palette for the line- and stripplot.return_fig (
bool, default:False) – if True, returns figure of the plot, that can be used for saving.
- Return type:
- Returns:
If return_fig is True, returns the figure, otherwise None.
Examples
>>> # Example with EdgeR >>> import pertpy as pt >>> adata = pt.dt.zhang_2021() >>> adata.layers["counts"] = adata.X.copy() >>> ps = pt.tl.PseudobulkSpace() >>> pdata = ps.compute( ... adata, ... target_col="Patient", ... groups_col="Cluster", ... layer_key="counts", ... mode="sum", ... min_cells=10, ... min_counts=1000, ... ) >>> edgr = pt.tl.EdgeR(pdata, design="~Efficacy+Treatment") >>> edgr.fit() >>> res_df = edgr.test_contrasts( ... edgr.contrast(column="Treatment", baseline="Chemo", group_to_compare="Anti-PD-L1+Chemo") ... ) >>> edgr.plot_paired(pdata, results_df=res_df, n_top_vars=8, groupby="Treatment", pairedby="Efficacy")
- Preview:
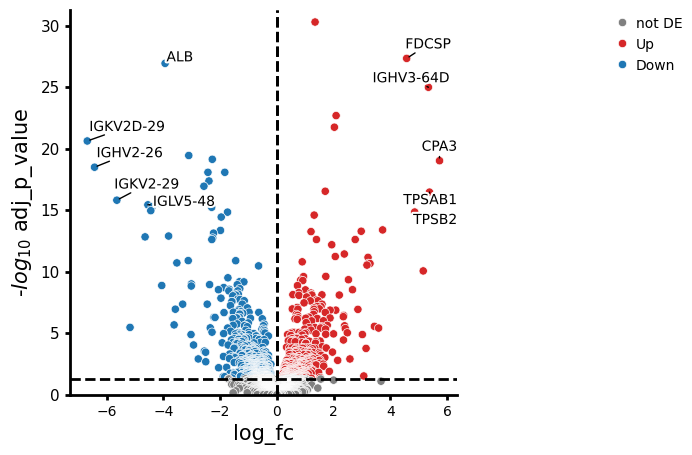
- PermutationTest.plot_volcano(data, *, log2fc_col='log_fc', pvalue_col='adj_p_value', symbol_col='variable', pval_thresh=0.05, log2fc_thresh=0.75, to_label=5, s_curve=False, colors=None, varm_key=None, color_dict=None, shape_dict=None, size_col=None, fontsize=10, top_right_frame=False, figsize=(5, 5), legend_pos=(1.6, 1), point_sizes=(15, 150), shapes=None, shape_order=None, x_label=None, y_label=None, return_fig=False, **kwargs)#
Creates a volcano plot from a pandas DataFrame or Anndata.
- Parameters:
log2fc_col (
str, default:'log_fc') – Column name of log2 Fold-Change values.pvalue_col (
str, default:'adj_p_value') – Column name of the p values.symbol_col (
str, default:'variable') – Column name of gene IDs.varm_key (
str|None, default:None) – Key in Anndata.varm slot to use for plotting if an Anndata object was passed.size_col (
str|None, default:None) – Column name to size points by.point_sizes (
tuple[int,int], default:(15, 150)) – Lower and upper bounds of point sizes.pval_thresh (
float, default:0.05) – Threshold p value for significance.log2fc_thresh (
float, default:0.75) – Threshold for log2 fold change significance.to_label (
int|list[str], default:5) – Number of top genes or list of genes to label.s_curve (
bool|None, default:False) – Whether to use a reciprocal threshold for up and down gene determination.color_dict (
dict[str,list[str]] |None, default:None) – Dictionary for coloring dots by categories.shape_dict (
dict[str,list[str]] |None, default:None) – Dictionary for shaping dots by categories.fontsize (
int, default:10) – Size of gene labels.colors (
list[str], default:None) – Colors for [non-DE, up, down] genes. Defaults to [‘gray’, ‘#D62728’, ‘#1F77B4’].top_right_frame (
bool, default:False) – Whether to show the top and right frame of the plot.figsize (
tuple[int,int], default:(5, 5)) – Size of the figure.legend_pos (
tuple[float,float], default:(1.6, 1)) – Position of the legend as determined by matplotlib.shapes (
list[str] |None, default:None) – List of matplotlib marker ids.shape_order (
list[str] |None, default:None) – Order of categories for shapes.return_fig (
bool, default:False) – if True, returns figure of the plot, that can be used for saving.**kwargs (
int) – Additional arguments for seaborn.scatterplot.
- Return type:
- Returns:
If return_fig is True, returns the figure, otherwise None.
Examples
>>> # Example with EdgeR >>> import pertpy as pt >>> adata = pt.dt.zhang_2021() >>> adata.layers["counts"] = adata.X.copy() >>> ps = pt.tl.PseudobulkSpace() >>> pdata = ps.compute( ... adata, ... target_col="Patient", ... groups_col="Cluster", ... layer_key="counts", ... mode="sum", ... min_cells=10, ... min_counts=1000, ... ) >>> edgr = pt.tl.EdgeR(pdata, design="~Efficacy+Treatment") >>> edgr.fit() >>> res_df = edgr.test_contrasts( ... edgr.contrast(column="Treatment", baseline="Chemo", group_to_compare="Anti-PD-L1+Chemo") ... ) >>> edgr.plot_volcano(res_df, log2fc_thresh=0)
- Preview: